


User Manual to “FMFilter: A Fast Model Based Variant Filtering Tool”
1- FMFilter Setup and PrerequisitesThe installation interface provides all necessary external
libraries for the program to run in Windows 32 or 64 bit and Linux
systems.
You can download the latest version of FMFilter from IGBAM web page: http://www.igbam.bilgem.tubitak.gov.tr/en/softwares/FMFilter.
You can follow the steps provided by the setup executable file to build FMFilter.
2- Input data format
2.1 Variant call format (*.vcf) file (required)
As an input file, FMFilter accepts *.vcf outputs from different variant identification tools such as GATK [1] which is a widely used program to process next-generation sequencing (NGS) data. The user does not need to do additional conversion steps. The details of .vcf file format can be found in 1000 Genome website [2]. A sample vcf file , which includes a family with two daughters affected by Mabry syndrome [3] used throughout this manual is provided in FMFilter website.
3-User Interface and Usage
3.1 Defining Annotation
To have an effective usage we recommend to define frequently used annotations using annotation finder tool. To open the annotation finder tool the user clicks the Tools > Annotation Finder menu at top-left of the program window depicted in Figure 1.

Figure 1. Annotation Finder
This operation will open an Annotation Finder window shown
below. The user could define and save the annotations that he will use
later following steps below.
Step1: Open the .vcf file from the Browse button. This brings the info field sections which includes in the annotation.
Step2: The related fields for gene name and mutation type must be chosen.
Step3: The preferred population frequencies that will be used in minor allele frequency (MAF) filtering must be chosen.
Step4: Press the button Find Type Keywords to list the type of mutations to the Keyword field that are included in the .vcf file.
Step5: The user can save this annotation with a preferred name that he/she will use later in filtering.
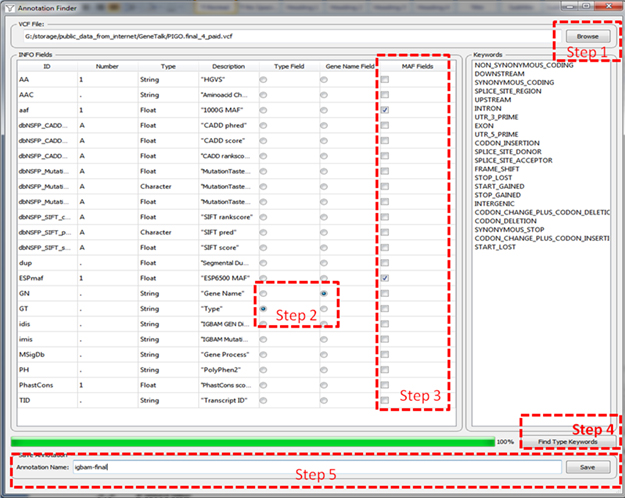
Figure 2. Steps of defining an initial annotation
3.2 Main Window and Usage of FMFilter
Figure 3 represents the general outline of FMFilter GUI. The detailed information for each region is provided below.
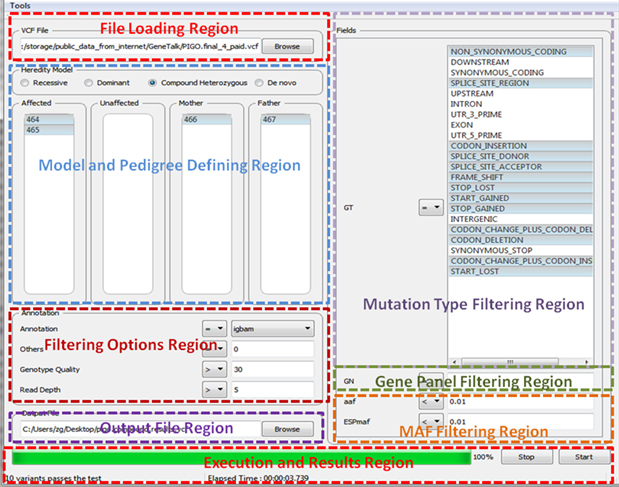
Figure 3. The general outline of FMFilter user interface.
3.2.1 File Loading
*.vcf file is uploaded from the selected file location as shown in File Loading Region of Figure 3 . After opening the .vcf file all the including samples are listed in Model and Pedigree Defining Region of Figure 3 .
3.2.2 Model and Pedigree DefiningThe user chooses the inherence model for further analysis. The type of pedigree information may change according to the model.
Recessive Model
The user can choose affected, unaffected individuals and the parents
according to the pedigree samples available using the window presented
in Figure 4 . In this model the mother and father are assumed to be
heterozygous carriers and the affected children are considered as
homozygous variant. If there exist unaffected siblings, they are taken
as either heterozygous carries or homozygous reference.

Figure 4. Pedigree Definition in the Recessive Model
3.2.2.1 Dominant ModelIn this model we assume that the affected individuals carry a single copy of a deleterious variant. Therefore we distinguish only the affected an unaffected individuals. The parentship relation does not provide extra information. The user has to choose affected and unaffected individuals as in Figure 5 .

3.2.2.2 Compound Heterozygous Model
To have a compound heterozygous mutation the affected individual must carry at least two heterozygous mutations in one particular gene one of which comes from the mother and the other from the father. To be able to the carry out compound heterozygous analysis, the input .vcf file must be annotated using the information gene name and the user should introduce the program where the gene name is defined in the info field of *.vcf as defined in Defining Annotation Section. The user can choose affected, unaffected individuals and the parents as in Figure 6. The information from one or two parents are required to work properly.

Figure 6. Pedigree Definition in the Compound Heterozygous Model
3.2.2.3 De novo Model
Novel mutations which has not been observed either in the mother or father are considered. The information from at least one of the parents is required to have a valid filtering. The user has to choose one affected individual and the other unaffected individuals and parents as in Figure 7.

Figure 7. Pedigree Definition in the De novo Model
3.2.3 Filtering Options Region
Filtering Options Region consist of four different options as in Figure 8.

Figure 8. Filtering Options Region
Using the Annotation selection field the user can choose the annotation which he has been previously defined in the Annotation Defining section.
The Others option provides a method to filter polymorphic variants by using the other unrelated individuals in the .vcf file. If the others field is set to zero, it is assumed that the variant is observed only at the individuals selected according to the pedigree. If the others field is set to a number other than zero, we allow exceptions to be observed at most as the number defined. Even if the population frequency is not available, this option enables the user to reduce the number of variants significantly.
The Genotype Quality and Read Depth options are necessary to filter out the variants which do not have enough read quality and coverage. NGS technology is prone to this type of errors therefore these options could be useful.
3.2.4 Filtering Options Region
Mutation Type Filtering window enables the user to restrict the related search to the specific types of mutations considered. The user can prefer to list the most significant loss of function mutations first and then enlarge the results if he/she likes. The list of mutation types is determined by the annotation chosen at the beginning of the analysis.

Figure 9. Choosing Mutation Types
3.2.5 Gene Panel Filtering Region
This window makes it possible to filter with respect to specific set of genes considered. The gene name must be written the same as it is written in the annotation. The program looks for an exact match. Multi gene names must be separated with comma without a space. To be able to use gene panel filtering, the related fields for gene name must be previously defined at the Annotation Defining step.
![]()
Figure 10. Gene Panel Filtering Region
3.2.6 MAF Filtering Region
To be able to use MAF filtering, the related fields for population frequency must be previously defined at the Annotation Defining step. Population frequency is an important feature for filtering variants. FMFilter supports to the filtering with respect to 1000G or ESP minor allele frequency (MAF) whenever the annotations of these databases are annotated in the .vcf file. FMFilter supports custom annotation therefore the user can use his in-house population statistic which would be much more powerful to eliminate false alarms than general population statistics.

Figure 11. MAF Filtering Region
3.2.7 Output File Region
The user is required to define the path and the name of the result file. Output file is generated in the .vcf format for further analysis.

Figure 12. Output File Region
3.2.8 Execution and Results Region
The user can start and stop the analysis with respect to options set in previous steps. He/she can see number of variants passed the filtering and the elapsed time after the execution. The green progress bar shows the percentage of the completed analysis.
![]()
Figure 13. Execution and Results Region
References:
1. McKenna, A., et al., The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 2010. 20(9): p. 1297-1303.
2. Altshuler, D., et al., A map of human genome variation from population-scale sequencing. Nature, 2010. 467(7319): p. 1061-1073.
3. Kamphans, T., et al., Filtering for Compound Heterozygous Sequence Variants in Non-Consanguineous Pedigrees. PLoS ONE, 2013. 8(8): p. e70151.